We are excited to announce a new batch of small molecule screening simulations are now up and running on Folding@home! These simulations will help prioritize which molecules will be synthesized and assayed by the COVID Moonshot aiming to rapidly developing new therapies against the SARS-CoV-2 main viral protease.
Please note that these new projects can only be run on CPUs. They complement the many Folding@home COVID-19 projects that are already running on GPUs, and they allow everyone to join in to help in the fight against COVID-19.
About the project
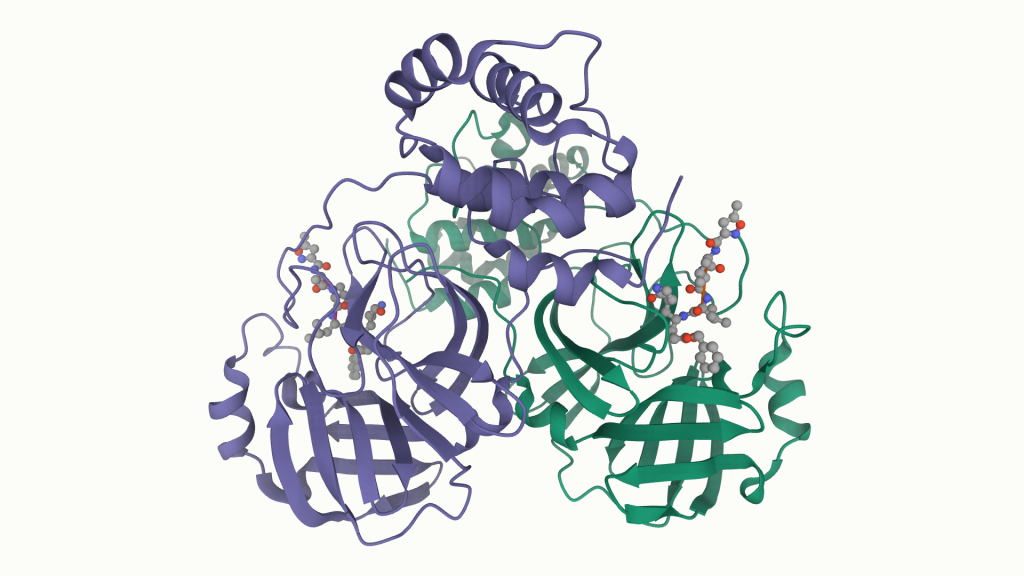
The Voelz and Chodera labs have been working with an international team of researchers to computationally screen potential inhibitors of theSARS-CoV-2 coronavirus main protease (also known as SARS-CoV-2 MPro or 3CL). After the virus gets into a cell, it co-opts the cell’s translational machinery and makes a polyprotein (one long chain of amino acids) that needs to be cleaved into smaller pieces that become the functional parts of the virus. The main protease is the viral protein that does the cleavage.
SARS-CoV-2 main viral protease (Mpro) (pdbid: 6LU7)
We are using an alchemical free energy calculation method called free energy perturbation, or FEP, to estimate the affinity of a potential drug molecule to its receptor. FEP methods are very expensive compared to other methods like docking, but with the advantages of high accuracy and physical insight. We are using protocols very similar to those used in the most recent SAMPL blind challenge.
The calculation involves decoupling the drug molecule (by turning off its electrostatic and dispersion interactions) from its receptor, and again for a molecule in water. For the latter type of simulations, unfortunately there’s not much to look at the Folding@home viewer, because there’s no protein being simulated. However boring these projects appear, please keep in mind that they are absolutely essential to our success.


 发表于 2020-3-31 21:07:05
发表于 2020-3-31 21:07:05
 发表于 2020-4-2 22:34:19
发表于 2020-4-2 22:34:19