|
|
 |

FightAIDS@Home |
|
- What is FightAIDS@Home?
- How do I join the FightAIDS@Home Project?
- How does the FightAIDS@Home software work?
- Will my computer only be working on the
FightAIDS@Home project?
- What computers can run FightAIDS@Home?
- Panel A: Current Dockings
- What are the colored spheres in Panel A?
- Panel B: Docking Energies
- What is electrostatic energy?
- What is non-bonded energy?
- Panel C: Best Docking Energy
- Current Progress Bar
What is FightAIDS@Home?
FightAIDS@Home is a project of the Olson
laboratory that uses distributed computing to contribute your
computer's idle resources to accelerate research into new drug
therapies for HIV, the virus that causes AIDS.
How do I join the
FightAIDS@Home Project?
All you need to do to join FightAIDS@Home is download and install
the free
software. Once that has been done, your computer is then
automatically put to work and you continue using your computer as
usual.
How does the
FightAIDS@Home software work?
We use software that automatically downloads small pieces of data
and performs calculations that model how drugs interact with
various HIV virus mutations. After your computer processes the
information, the results are sent by World Community Grid to The
Scripps Research Institute where they are analyzed by the Scripps
research team. The process takes an enormous amount of computing
time, which is why The World Community Grid needs you (and your
friends!) to participate in FightAIDS@Home.
Will my computer only
be working on the FightAIDS@Home project?
Your computer will work on whatever projects you want. You can
select from the projects currently active at World Community Grid
by visiting the My
Projects page. There you can view all available projects, and
choose those in which you want to participate. (Currently, only
Windows computers have the ability to opt-in or out of specific
projects.)
What computers can run
FightAIDS@Home?
Currently, the system requirements for this project are:
- Connection to the Internet
- A Windows PC with a Pentium 550MHz or faster processor
- 250 MB of RAM or more
- 50MB of free hard disk space
 |
Panel A: Current
Dockings
Click on the  on your agent application window in the lower right hand corner.
You then will see a graphics window similar to the following:
on your agent application window in the lower right hand corner.
You then will see a graphics window similar to the following:
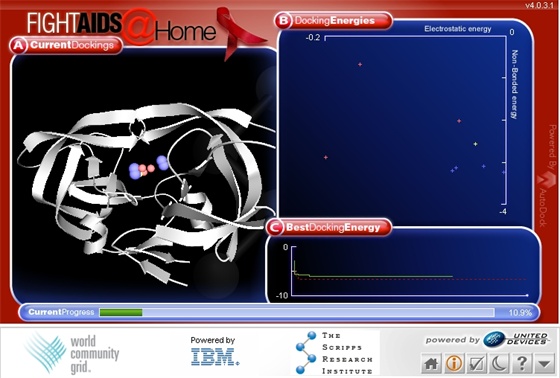
What is the white arrow, helix and
loopy structure?
Ribbon diagrams are simplified drawings of proteins that make it
easier for scientists to view and understand what is shape is. The
three-dimensional "skeleton" of HIV-1 protease is shown
as a white ribbon diagram on the screen and is magnified about
10,000,000 times.
In this panel, you can see the shape that the particular sequence
of amino acids in HIV-1 protease makes in three dimensions. For
clarity, we are not showing the details of all of the atoms in the
protein molecule, just the backbone. Remember, all proteins,
including HIV-1 protease, are made up of strings of amino acids,
linked like beads on a string. There are twenty different
naturally-occurring amino acids, and you can think of them as
different kinds of building blocks. These strings of amino acids
have parts that like to stick to others while repelling others.
The different parts of the protein's amino acid chain clump
together into characteristic three-dimensional shapes.
|
What are the colored
spheres in Panel A?
The search algorithm used in AutoDock is not just looking at one
possible solution of one candidate drug molecule (ligand) but is
actually evaluating many possible solutions at once. The spheres
show places where the best drug molecule to HIV-1 protease
dockings have been calculated and the color shows how good they
are.
AutoDock is trying to find the best way that the current ligand,
the one your agent has downloaded, can fit together with the
target HIV-1 protease. You can think of the ideal drug we are
trying to find as a "key," and the HIV-1 protease as a
"lock." Unlike keys in the real world, however, many
drug molecules bend to change shape. In this respect, molecules
are like a dancer's body; the same body is able to adopt many
different poses and shapes. Unfortunately, we do not know what
shape a candidate drug will adopt until we try millions of
different possibilities and then select the best one.
To find the best fit, we are using an algorithm. An algorithm is
just a recipe, a list of ingredients and instructions on how to do
or make something. We are actually applying the principles of
evolution in our search algorithm to find the best way that our
candidate drug molecule would best fit together with the target,
HIV-1 protease. Like evolution in the real world, we have a
"population" of possible solutions to the problem.
This is what you are seeing when you look at the different colored
spheres dotted around the white ribbon diagram. The colors
correspond to the same colors of the crosses in panel B. Those
representing more negative energy are considered better dockings.
AutoDock uses a representation for each of these ligand dockings
that says where the ligand's center is, what its orientation is,
and what shape it has currently adopted. AutoDock applies genetic
operations on the representations of random pairs of ligand shapes
to generate two new representations and hence potentially better
solutions. You can see how well AutoDock is doing by looking at
the graph in panel C.
|
Panel B: Docking
Energies
We see here the energy breakdown for each candidate ligand docking
of the current population of possible solutions. The total energy
of a ligand binding to the HIV-1 protease consists of an
electrostatic energy component and a non-bonded energy component.
The electrostatic energy measures how many like-charges and
unlike-charges are interacting between the ligand and the
protease. The non-bonded energy measures non-electrostatic
attraction between the two.
|
What is electrostatic
energy?
You can see electrostatic forces in action if you rub a balloon on
a dry wooly sweater, and then gently place the balloon against a
wall: It sticks! This is because all objects are made of atoms.
Each atom has an equal number of electrons and protons. Electrons
have a negative charge, while protons have a positive charge.
These charges balance one another exactly to make objects neutral,
or uncharged. When we rub the balloon against a sweater, the
friction causes electrons to be rubbed off the sweater and onto
the balloon. The balloon becomes charged with static electricity,
and it now has more electrons than protons, so it is negatively
charged; the wall is more positively charged than the balloon so
the balloon sticks.
If you were to rub a second balloon on your sweater, and hang the
two balloons from a string, you would see the two balloons repel
one another.
|
What is non-bonded
energy?
Non-bonded energy arises because atoms are "sticky" when
they get close to one another. The amount of
"stickiness" depends on the two atoms that are
interacting. However, atoms repel one another when they are pushed
too close together. Between two touching molecules, there are many
of these non-bonded interactions. They are called
"non-bonded" because these interactions are not
permanent like chemical bonds.
|
Panel C: Best Docking
Energy
We see here the best docking energy in the current population,
plotted over the course of the current docking, shown as a green
solid line. The red-dotted line shows the same kind of graph, but
for the best docking achieved so far. As the current docking
proceeds, at the end of every generation, the green graph gets
updated.
The vertical axis shows the best energy. The more negative the
energy, the better, i.e. the more precisely we predict this
particular ligand will bind to the protease. You can see times
when the energy is not changing (the horizontal lines in the
graph) and times when the energy dropped (the vertical lines) when
AutoDock has found a better solution than the previous generation.
|
Current Progress Bar
The Current Progress Bar shows how much of the current work unit
has been completed. The work units are specified by the
researchers at The Scripps Research Institute and transmitted via
the servers at World Community Grid to your machine. Each work
unit has just one candidate drug molecule, out of a vast library
of candidate drug molecules we are virtually screening. The
software running under the grid agent on your computer is called
AutoDock, and it tries to determine the best way the current
ligand fits into the target HIV- 1 Protease. When the work unit is
finished, the best results are sent back to Scripps via World
Community Grid for further analysis, to find the best candidate
protease inhibitors for further testing in the laboratory.
|
